Ph.D. Thesis
Title Synthesis and Reactivity of Metalloporphyrins
in (A) Biomimetic Studies of Terminal Oxidases and (B) the Preparation
of Novel Heterodinuclear Multiple Metal-Metal Bonds.
Adviser Prof. James P. Collman
Thesis Committee Prof. Gordon Braun, Dep. of Geology; Prof.
Edward Solomon, Dep. of Chemistry; Prof. Chris E. D. Chidsey, Dep.
of Chemistry; and Prof. T. Daniel P. Stack, Dep. of Chemistry
Essay
The unique properties of porphyrins as transition metal
ligands underlie their prevalence as cofactors in the biosphere and
also make them valuable components of artificial chemical systems
based on d-block elements. My Ph. D. work in the Collman lab at Stanford
used porphyrins in both contexts. For example, I utilized porphyrins
to stabilize and isolate a MoRe5+ core [ref. 1], which
contains the first ever quadruple bond between elements from different
groups [ref. 2]. I also studied monomeric RhII porphyrins,
which cleave C-H bonds in methane and other hydrocarbons. Yet to be
useful as hydrocarbon activation catalysts, the RhII center
must survive in the presence of nucleophiles: something that was thought
to be impossible before I realized it in a judicially chosen nucleophile/porphyrin
system [ref. 3]. But most of my Ph. D. research was devoted to biomimetic
studies of the O2-reduction site in terminal oxidases.
Heme/Cu terminal oxidases (e.g., cytochrome oxidase, CcO) make possible
terrestrial life as we know it. At their bimetallic heme/Cu site (Figure
1A) these enzymes catalyze the final step in the respiratory electron
transfer chain: 4-electron, 4-proton reduction of O2 to
H2O. This highly exergonic reaction is coupled to proton
translocation against the transmembrane electrochemical gradient,
which drives ATP synthesis. Because terminal oxidases are so complex
and their isolations are often heterogeneous, biomimetic work can
invaluably complement biochemical studies of the enzyme. I chose biomimetic
chemistry to understand the possible roles of the distal Cu ion in
steady-state O2 reduction by terminal oxidases. This question
is one of the more contentious issues of these enzymes’ biochemistry
since it can not be probed by studying terminal oxidases themselves
and prior biomimetic studies lead to several contradicting hypotheses
[ref. 4].
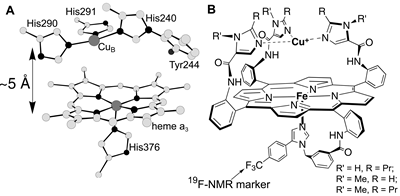
Figure
1. The O2 reducing site of cytochrome oxidase (A)
and the biomimetic analogs synthesized and studied as a part of my
Ph.D. work (B).
To arrive at biologically-relevant conclusions a biomimetic
chemist must synthesize a molecule that faithfully reproduces both
the stereoelectronic properties and the reactivity of the target enzymatic
site, and study its properties under conditions as close as possible
to the ones under which the enzyme operates. I synthesized a series
of metalloporphyrin complexes that are the closest structural analogs
of the heme/Cu site ever reported (Figure 1) [refs. 5,4] and established
that these biomimetic catalysts reproduce the catalytic reactivity
of the heme/Cu site toward O2 and H2O2
[refs. 6,7,8]. My catalysts reduce O2 at physiologically-relevant
pH and electrochemical potentials,
with high selectivity (>98%) and stability (>104
turnovers), and at maximum rates comparable to those reported for
CcO. This catalysis proceeds via turnover-limiting generation of a
formally ferric-hydroperoxo intermediate, pFeIII-OOH (Figure
2). This is significant because the most likely source of H2O2
in metalloporphyrin-catalyzed O2 reduction is hydrolysis
of pFeIII-OOH [ref. 4]. Because the steady-state concentration
of the intermediate produced in the turnover-determining step is minimal,
the flux of H2O2 is minimized as well [ref.6].
Indeed, reduction of O2 by cytochrome oxidase is thought
to proceed analogously: a slow generation of the ferric-hydroperoxo
intermediate followed by its
rapid reduction.
In order the study catalytic O2 reduction
under physiologically-relevant diffusion-limited electron flux, I
developed a system wherein isolated mobile catalyst molecules are
dispersed in a lipid matrix at the electrode surface [ref. 7]. In
contrast to the electrode-adsorbed FeCu and Cu-free catalysts, which
catalyze O2 reduction under fast electron flux and manifest
similar kinetics, mechanism and stability (Figure 2), with slow electron
flux the FeCu catalyst is notably superior to the Cu-free analog,
displaying higher stability, turnover frequency and selectivity. These
findings suggest that the puzzling inactivity of the CuB-free mutants
of CcO may be due to rapid degradation of the catalytic site rather
than an intrinsic inability of the heme alone to reduce O2.
In the absence of an intramolecular electron source, such as CuI
, reduction of O2 by the Cu-free catalyst results in oxidation
of the porphyrin or imidazoles to chemically-unstable cation-radicals,
thereby leading to rapid catalyst degradation and loss of activity.
The totality of my findings suggests that in a stereoelectronic
environment similar to that of the heme/Cu site, the distal Cu ion
is an electron storage site. Apparently, O2 reduction
at this bimetallic site requires an activation energy that is not
significantly lower than that needed at the heme alone, provided electrons
are available from elsewhere.
While Cu may seem to be just a “spectator”
in O2 reduction under the excess of electrons, it has pronounced
physiological benefits by modulating the heme’s reactivity. Without
affecting
the kinetics or the mechanism of O2 reduction in this regime,
distal Cu decreases the amount of toxic partially-reduced oxygen byproducts
generated by the O2-reducing catalyst, by suppressing superoxide-releasing
autooxidation of oxyheme, (por)FeO2 [ref. 6]. Cu also makes
the FeCu catalyst less susceptible to CO and CN- inhibition
[ref. 8], particularly at physiologically-relevant potentials. CN-
is cytotoxic because CN-ligated oxidized CcO can not be reduced in
vivo, which shuts down respiration and leads to death. My studies
suggest that with only a monometallic catalytic site at CcO, 5-times
lower concentrations of CN- would cause such shutdown.
Cu decreases the susceptibility by increasing the FeIII/II
potential and by destabilizing (probably sterically) CN-
binding to both the reduced (FeIICuI) and (unexpectedly!)
oxidized (FeIIICuII) catalysts.
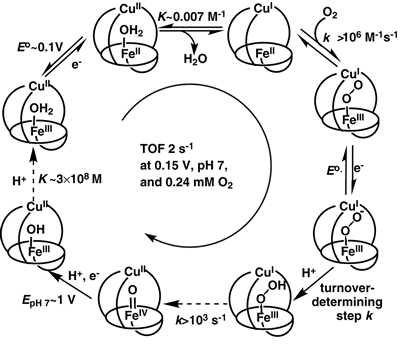
Figure
2. The kinetic mechanism of catalytic O2 reduction by the
biomimetic CcO analogs under physiologically-relevant conditions and
rapid electron flux, when Cu does not affect the mechanism. Kinetics
and/or thermodynamics of every step other than those generating the
pFeO2- and pFeO2H intermediates were
studied independently. The broken arrows indicate essentially irreversible
steps, which do not affect the overall turnover frequency (TOF).
I was fortunate to work with biomimetic catalysts that
are unique in how closely they reproduce both the structure and some
key reactivity of CcO. Such fidelity makes it much more likely that
the effects of Cu observed in my studies, however unexpected, also
influence the reactivity of CcO. More generally, my work has demonstrated
that biomimetic studies of a complex enzyme can yield a well-supported
and self-consistent hypothesis regarding an aspect of enzymatic reactivity
that could not be unraveled by studying the enzyme itself. This work
also extends our fundamental understanding of an important but deceptively
simple reaction (reduction of O2 to H2O) by
demonstrating facile heterolysis of the O-O bond in H2O2
and its
derivatives by a molecular catalyst. This undermines the dominant
paradigm in the field of catalytic O2 reduction that O-O
bond heterolysis is intrinsically the most difficult part of the O2
reduction cycle. Finally, the techniques I developed, such as electrode-confined
lipid films to model diffusion-limited electron fluxes in biological
environments and the use of O2- and H2O2
scavengers to identify primary partially-reduced oxygen byproducts,
should be valuable to other areas of chemistry.
References
1. Collman, J. P.; Boulatov, R.; Jameson, G. B. The First Quadruple
Bond Between Elements of Different Triads. Angew. Chem. Int. Ed.
2001, 40, 1271–1274.
2. Collman, J. P.; Boulatov, R. Heterodinuclear Transition Metal Complexes
with Multiple Metal-Metal Bonds. Angew. Chem. Int. Ed. 2002,
41, 3948-3961.
3. Collman, J. P.; Boulatov, R. Synthesis and Reactivity of Porphyrinatorhodium(II)-Triethylphosphine
Adducts: The Role of PEt3 in Stabilizing a Formal Rh(II)
State. J. Am. Chem. Soc. 2000, 122, 11812-11821.
4. Collman, J. P.; Boulatov, R.; Sunderland, C. J. Functional and
Structural Analogs of the Dioxygen-Reduction Site in Terminal Oxidases.
In The Porphyrin Handbook; Kadish, K. M.; Smith, K. M.; Guilard,
R.; Eds. Academic Press: 2003, Vol. 11, pp. 1-49.
5. Collman, J. P.;. Sunderland, C. J.; Boulatov, R Biomimetic Studies
of Terminal Oxidases: Trisimidazole Picket Metalloporphyrins. Inorg.
Chem. 2002, 41, 2282-2291.
6. Boulatov, R.; Collman, J. P.; Shiryaeva, I.; Sunderland, C. J.
Functional Analogs of the Dioxygen-Reduction Site in Cytochrome Oxidase:
Mechanistic Aspects and Possible Effects of CuB. J.
Am. Chem. Soc. 2002, 124, 11923-11935.
7. Collman, J. P.; Boulatov, R. Electrocatalytic Dioxygen Reduction
by Synthetic Analogs of the Heme/Cu Site of Cytochrome Oxidase Incorporated
in a Lipid Film. Angew. Chem. Int. Ed. 2002, 41, 3487-3489.
8. Collman, J. P.; Boulatov, R.; Shiryaeva, I.; Sunderland, C. J.
Distal Cu Ion Protects Synthetic Heme/Cu Analogs of Cytochrome Oxidase
against Inhibition by CO and Cyanide. Angew. Chem. Int. Ed.
2002, 41, 4139-4142.
> Back to Prize index
page

