|
|
Vol.
35 No. 2
March-April 2013
by Krishna N. Ganesh
The DNA double helix made up of two antiparallel strands complementary to each other through specific base pairing of A with T and G with C codes the key information necessary for synthesis and regulation of all proteins and enzymes required of functioning of a cell from cell division to cell differentiation and cell development. The discovery also gave birth to the fields of molecular and structural biology, which have been key to the genetic revolution that has resulted in the development of vital products, ranging from hormones and enzymes to therapeutic molecules and vaccines. The penultimate achievement stemming from the discovery of DNA’s structure was the unraveling of the entire human genome in 2001 and work continues unabated on the genomes of other organisms. These discoveries have implications for our understanding of diseases at the molecular level and, thereby, the development of cures. Some of the most spectacular applications take advantage of the self-assembling properties of the genetic molecule DNA to make nonbiological novel generic materials.
Chemistry has played a central role in the success of these processes, since easy availability of well-defined sequences of DNA in reasonable amounts has been crucial to most of these applications. Har Gobind Khorana’s saga of the first synthesis of a gene for Phe t-RNA was the first illustration of the power of chemistry in delineating biological processes. Crucial to the success was the development of effective and compatible protecting groups for the exocyclic amino groups of the nucleobases and the hydroxyls of ribose moiety. Since then, it has inspired generations of chemists to improve, optimize, and invent new methods of facile synthesis. The pioneering work of Robert Letsinger and Marvin Caruthers has enabled the synthesis of DNA and RNA even by nonhumans (Automatic synthesis machines). The availability of chemically synthesized, well-defined sequences of oligonucleotides had a major impact on the acquisition of functional information about DNA structure and its interaction with a variety of molecules ranging from metal ions to small molecule drugs and regulatory proteins. Although the structure of DNA was proposed in 1953, it took almost 25 years for the first atomic structure of DNA to be solved, which completely validated the structure proposed by Watson and Crick. Then came the discovery of polymorphism in DNA (A, B, and left-handed Z forms), sequence-specific molecular recognition of DNA by RNA (DNA-RNA hybrids), intercalating and groove-binding molecules, restriction enzymes, repressor proteins, zinc finger proteins, and the structure of DNA triplexes and tetraplexes. Synthetic oligonucleotides as primers were central to Kary Mullis’ discovery of polymerase chain reactions and Sanger’s DNA sequencing method, which eventually made the human genome sequencing possible. Gene cloning was made possible by the availability of synthetic oligonucleotides as linkers to generate suitable fragments for pasting of DNA fragments in the desired way. The evolution of SELEX as a technique to discover unknown interactions of DNA with proteins and ligands, hinges on the capability of simultaneous synthesis of millions of oligonucleotides in a combinatorial way. This has led to the discovery of aptamers, ribozymes, and DNAzymes, from which a number of practical applications have already emerged.
When everyone thought that DNA synthesis has reached saturation in terms of chemistry, came the concept of antisense therapeutics and ribozymes in 1990s, which expanded the synthesis scope to chemically modified nucleic acids. Here, a short stretch of DNA inhibits protein synthesis by complementation-steric blocking of m-RNA reading by ribosomes. Since oligonucleotides reluctantly enter cells due to their anionic character and have short half-lives within the cells (nuclease susceptible), they have to be structurally modified to overcome the above lacunae. This expanded organic synthesis to a wide variety of structural analogues of DNA and RNA. One of the analogues (phosphorothioates) was approved as a drug (Vitravene) for retinosis in 1997 and just recently (29 January 2013) the second drug Mipomersen was approved by the U.S. FDA for a rare cholesterol disorder. The recent discovery of siRNA and miRNA as efficient gene expression probes and, hence, potential therapeutic agents, has pushed the synthetic scope to newer limits. Contrary to initial apprehensions of scaling up synthesis of oligonucleotides, technology has evolved for large-scale production of multikilogram amounts of DNA and its analogues, which has made possible the successful clinical trials. Quite a few more DNA/RNA based drugs are in advanced stages of clinical trials, close to approval for several cancer and viral diseases.
The recent progress in DNA origami pioneered by Ned Seeman and DNA nanotechnology by Chad Mirkhin based on perfect self-assembling properties of complementary DNA strands is bringing a sea revolution to the application of DNA as a generic material. The ability to manipulate and program DNA in engineered cells has given birth to synthetic biology, a logical next step to genetic engineering with potential for artificial synthesis of a functioning cell. The sky seems to be the limit for such unabated progress in the application of chemistry to understand and even direct biology in new directions.
Looking back, the genius of Watson and Crick is still awe-inspiring. The elucidation of DNA’s structure followed by the creative contributions of innumerable chemists over the past 60 years has resulted in methods for transforming the genetic material in DNA into the generic “materials and drugs of the future.”
Krishna N Ganesh <[email protected]> is president of the IUPAC Organic and Biomolecular Chemistry Division.
by Marvin Caruthers
When I attended my first Nucleic Acids Gordon Conference in 1975, I was asked to review the state-of-the-art in oligonucleotide synthesis. At that time in my young career at the University of Colorado, we were focused not on DNA chemical synthesis, but on determining how proteins recognize nucleic acids. This was five years before we developed the phosphoramidite chemistry for DNA synthesis. Because I had established a reputation in nucleic acid synthesis through my graduate and postdoctoral research, it was only natural for me to present this lecture. During the evening meal following my review, a molecular biology attendee, who is now a member of the U.S. National Academy of Science, asked me “Marv, why do you want to learn how to chemically synthesize DNA? Certainly Khorana used synthetic DNA to solve the genetic code and now he has synthesized a gene. But what else can you do with it? Surely, you can find something more exciting to do.” This lack of interest in DNA synthesis among biologists, biochemists, molecular biologists, and geneticists was very common at that time. They couldn’t care less about the field.
There were a few laboratories that understood the power of synthetic DNA and how it could be used in biology. For example, we were chemically synthesizing the lac, cI, and cro operators for studies focused on how proteins recognize DNA.1 Others were synthesizing genes for human growth hormone2 and insulin3 which became the first genetically engineered, commercial therapeutic products, and Sanger’s laboratory was using synthetic DNA to develop a methodology for sequencing DNA.4 However, only a very few laboratories were focused on developing new procedures for chemically synthesizing DNA. At this time, only two methods were available—the phosphodiester and phosphotriester approaches. Unfortunately, these synthetic strategies were not accessible to biologists—the scientists who could use DNA in their research. This was because the procedures were complicated, time consuming, and required a trained chemist. Since this brief review cannot be comprehensive, I will only outline the chemistry we developed between 1976–1980 that has become known as the phosphoramidite methodology. Although introduced over 30 years ago, it remains the standard for oligonucleotide synthesis today.
Early observations from Robert Letsinger’s laboratory5 led us to explore using 2’-deoxynucleoside P(III) derivatives for synthesizing 2’-deoxyoligonucleotides on polymeric supports. Our research was predicated on the use of HPLC-grade (high-performance liquid chromatography) silica as a support for several reasons. One was that previous research on organic polymers had failed in part because these materials readily adsorbed the synthons and, thus, their removal after each condensation step was very difficult. This problem, when coupled with the knowledge that HPLC-grade silica had been designed for efficient mass transfer, dictated that it should be explored. Other reasons were that silica would be inert towards reactions with all the reagents we contemplated using and that it was a rigid, non-swelling matrix in common organic solvents. Therefore, HPLC-grade silica could be packed into a column and reactants merely pumped through the column.
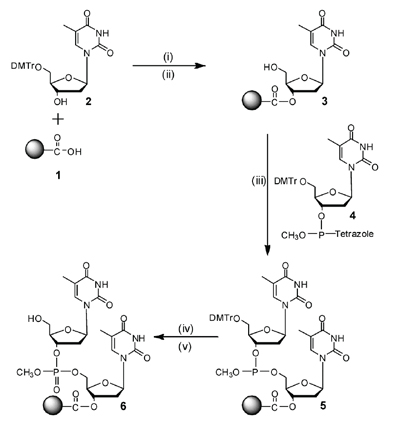 |
| Figure 1. Synthesis of DNA on Silica Supports Using 2'-Deoxynucleoside Phosphites. |
The initial approach we developed6 is outlined in figure 1. The first step was activating the silica matrix by attaching (3-aminopropyl)triethoxy silane to silica and then treating the product of this reaction with succinic anhydride to generate 1. The next step (i) was attaching 5’-dimethoxytrityl 2’-deoxythymidine to the support using dicyclohexylcarbodiimide to activate the carboxylic acid for condensation with the 2’-deoxynucleoside. After removal of the 5’-dimethoxytrityl group with acid (ii) to yield 3, synthesis with an activated 2’-deoxynucleoside 3’-phosphite was then carried out (iii). Of those we examined, the most reactive, while generating the fewest side-products, were the 5’-dimethoxytrityl 3’-methyltetrazoyl phosphites, 4. Using these synthons, 95 percent yields per condensation were observed during synthesis of the phosphite triesters 5. After oxidation of the phosphite to phosphate with aqueous iodine (iv) followed by removal of the dimethoxytrityl group with acid (v) to yield 6, the cycle could be repeated numerous times to generate a deoxyoligonucleotide. Finally, after completion of the deoxyoligonucleotide synthesis, the methyl group was removed from phosphate using a thiol. The oligomer was then cleaved from the support and base protecting groups (N-isobutyroylguanine, N-benzoylcytosine, and N-benzoyladenine) removed with ammonium hydroxide. Based upon the amount of dimethoxytrityl cation released following synthesis of a 12mer having all four bases, the overall yield was 55 percent, which at that time was unprecedented in the nucleic acid field. During the course of this work, we also developed a semiautomatic machine where one cycle of synthesis on a silica column, including all reagents and solvents, could be programmed and completed automatically. The operator added the next appropriately protected 2’-deoxynucleoside 3’-phosphite to the column and initiated another synthesis cycle. Although more successful than any previous method, it was far from acceptable. The main problem was that the 2’-deoxynucleoside 3’-tetrazoylphosphites had to be prepared at -78° C and preferably used the same day. These are procedures that are not appropriate for biologists and biochemists. Moreover, a phosphite adduct on the N-isobutyroylguanine base was very stable and could be removed only by several treatments with anhydrous pyridine—again a procedure unacceptable for routine synthesis. If these adducts were not removed, yields of deoxyoligonucleotides containing 2’-deoxyguanosine were very low. The major advantage of this approach, even to the present, was the successful development of silica as a matrix for DNA synthesis.
The final, major step in the development of the phosphoramidite methodology was discovering the 2’-deoxynucleoside 3’-phosphoramidites as synthons.7 This study was initiated with aminophosphines. The strategy was to synthesize 5’-dimethoxytrityl 2’-deoxynucleoside 3’-aminophosphines and then activate these synthons via insertion of CO2 , CS2 , or COS8,9 to form a mixed anhydride. These synthons would then be condensed with a 2’-deoxynucleoside. In our hands, this strategy was not successful. However during the attempted synthesis of these aminophosphines by reacting 5’-dimethoxytritylthymidine with N,N-dimethylaminomethoxychlorophosphine in pyridine, we observed on thin-layer chromatography the conversion of the 2’-deoxynucleoside to a dimethoxytrityl positive spot that moved similar to a dinucleotide. Based upon the literature, where there is good evidence for acid activation of aminophosphines,10 we reasoned that the initial product, 5’-dimethoxytrityl 2’-deoxythymidine 3’-N,N-dimethylaminomethoxyphosphine (a phosphoramidite), was further activated by pyridine hydrochloride, which was present in the reaction mixture due to the condensation of the chlorophosphine with the 2’-deoxynucleoside. This protonated 5’-dimethoxytrityl 2’-deoxythymidine 3’-O-methoxy-N,N-dimethylammonium phosphine would then react with excess 5’-dimethoxytrityl 2’-deoxythymidine present in the reaction mixture to form bis(5’-dimethoxytrityl 2’-deoxythymidine) 3’-O-methylphosphite.
The next challenge was to learn how to stabilize the 5’-dimethoxytrityl 2’-deoxythymidine 3’-phosphoramidite intermediates so they could be isolated, stored, and then activated during a controlled DNA synthesis. This was accomplished with the aid of phosphorus NMR and by using Hunig’s base to remove acid during aqueous work-up of reaction mixtures. Following isolation of these appropriately protected synthons, we immediately discovered that they were easily activated for DNA synthesis with anhydrous pyridine hydrochloride, other amine hydrochlorides, sulfonic acids, halogenated acetic acids, and even 2-nitropropane. However, for routine use by nonchemists, none of these acids was acceptable as they were hygroscopic or mutagenic. We therefore chose to recommend tetrazole as an acid activator because it could be readily purified by sublimation, stored as an anhydrous solid, maintained for days in anhydrous acetonitrile, and then used to synthesize DNA.
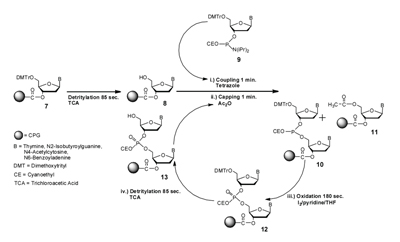 |
| Figure 2. The Phosphoramidite Approach for Synthesizing Oligonucleotides. |
These key developments led to the synthetic strategy as outlined in figure 2.11 The first step is conversion of the 5’-dimethoxytrityl 2’-deoxynucleoside linked to a silica support through a succinate ester to the corresponding 2’-deoxynuceloside by treatment with 2 percent trichloroacetic acid in dichloromethane to yield 8. The product of this detritylation is reacted (i) with tetrazole and 9, an appropriately protected 5’-dimethoxytrityl 2’-deoxynucleoside 3’-phosphoramidite to form 10, a phosphite triester. The next step is acylation with acetic anhydride in pyridine (ii) to generate 11. This step blocks any unreacted 2’-deoxynucleoside from further reaction and also removes phosphite adducts from the bases. The final steps in the cycle are oxidation of the phosphite triester to phosphate (iii) to yield compound 12 followed by detritylation with trichloroacetic acid (iv) to yield 13. Oxidation to phosphate is important as phosphite triesters are unstable to the trichloroacetic acid used in the detritylation step. The cycle is completed in approximately 5–7 minutes and can be used to easily prepare deoxyoligonucleotides 20–60 in length using both manual procedures and instruments.
For many years, the synthesis approach as outlined in figure 2 proved to be satisfactory for biological research where segments 20 or so oligomers in length were adequate. However, over the past 10 years, many new applications required DNA up to 300 oligomers in length. This development led me to collaborate with Emily LeProust and others at Agilent in order to design a synthesis protocol that could lead to DNA of this size.12 We now have a protocol where 250 000 unique DNA segments are prepared on a glass chip using a modified ink jet printer to deposit the activated 2’-deoxynucleoside phosphoramidites on the surface of this chip. Agilent currently has several DNA synthesizers operating 24/7 producing oligomers 250–300 in length. These machines each day produce the equivalent of the human genome (3 billion base pairs or 6 billion chemical couplings).
The phosphoramidite methodology has also been used to generate large numbers of DNA and RNA analogs with many finding applications in biology, therapeutics, and diagnostics. All were prepared in the Arbusov tradition using P(III) phosphorus derivatives of oligonucleotides to deliver P(V) analogs. For example, the sulfur containing DNA analogs, thiophosphate13 and dithiophosphate14 were prepared by sulfur oxidation of phosphites and thiophosphites respectively. By oxidation of phosphites with borane, borane phosphonate DNA has been recently synthesized15 and shown to be biologically active16 and to reduce metals.17 Using classical Arbusov-type chemistry, certain intermediate P(III) DNA derivatives can be converted to phosphate, phosphate triesters, phosphoramidates, and phosphorimidamidates.18 Moreover, other analogs derived from alkylaminophosphines can be converted to DNA having methylphosphonate,19 phosphonoacetate,20 phosphonoformate,21 and methylboranephosphine22 internucleotide linkages. This brief discussion on analogs derived from P(III) intermediates is far from complete, even for DNA, and only serves to introduce the possibility for applications in many other areas such as RNA, peptides, and oligosacharides.
The phosphoramidite approach has also been used extensively for the synthesis of RNA. However, the challenge in this field is identifying a series of orthogonal protecting groups for the 2’- and 5’- hydroxyls, phosphorus, and nucleoside bases. Although a large number of strategies have been tested, a recent approach utilizing 2’-thionocarbamates appears to be superior to others as higher cyclic yields and fewer side-products are observed while maintaining a rapid synthesis cycle and a high-yielding methodology for preparing the RNA synthons.23
The discovery of the phosphoramidite approach for DNA synthesis has been key to the development of several extremely important procedures commonly used in biology, biochemistry, and molecular biology. These include the use of polymerase chain reaction, DNA sequencing, and site-specific mutagenesis. When this DNA synthesis strategy was combined with other core procedures developed in the late 1970s to early 1980s (restriction modification of DNA; cloning of heterologous DNA elements into plasmids, phages, and chromosomes; and rapid protein sequencing), these new, very powerful technologies reinvigorated biological research and gave birth to the biotechnology industry.
Marvin H. Caruthers is a distinguished professor at the University of Colorado at Boulder. He has been a member of the National Academy of Sciences since 1994 and is a member of the American Academy of Arts & Sciences. He is the recipient of numerous awards, including the U.S. National Medal of Science in 2006, the Imbach-Townsend Award from the International Society of Nucleic Acid Research in 2006, the Promega Biotechnology Research Award in 2006, the National Academy of Sciences Award for Chemistry in Service to Society in 2005, and the Prelog Medal in Recognition of Pioneering Work on the Chemical Synthesis of DNA (ETH, Zurich, Switzerland) in 2004.
- D.V. Goeddel, et al., Proc. Natl. Acad. Sci. U.S.A 75, 3578–3582 (1978).
- D.V. Goeddel, et al., Nature 281, 544–548 (1979).
- D.V. Goeddel, et al., Proc. Natl. Acad. Sci. U.S.A 76, 106–110 (1979).
- F. Sanger, et al., Proc. Natl. Acad. Sci. U.S.A 70, 1209–1213 (1973).
- R.L.Letsinger and W.B. Lunsford, J. Am. Chem. Soc. 98, 3655–3661 (1976).
- M.D. Matteucci and M.H. Caruthers, J. Am. Chem. Soc. 103, 3185–3191 (1981).
- S.L. Beaucage and M.H. Caruthers, Tetrahedron Letters 22, 1859-1862 (1981).
- H.J. Vetter and H. Noeth, Chemische Berichte 96, 1308–1315 (1963).
- G. Oertel, et al., Chemische Berichte 97, 891–902 (1964).
- E.S. Batyeva, et al., in Chemistry of Organophosphorus Compounds, (Mir, Moscow, 1989), pp. 68–92.
- M.H. Caruthers, Science 230, 281–285 (1985).
- E.M. Leproust, et al., Nucleic Acids Res. 38, 2522–2540 (2010).
- W.J. Stec, et al., J. Am. Chem. Soc. 106, 6077–6079 (1984).
- W.K.D. Brill, et al., J. Am. Chem. Soc. 111, 2321–2322 (1989).
- H.B. McCuen, et al., J. Am. Chem. Soc. 128, 8138–8139 (2006).
- M. Olesiak, et al, Phosphorus, Sulfur, and Silicon and the Related Elements 186, 921–932 (2011).
- S. Roy, et al., Org. Biomol. Chem 10, 9130–9133 (2012).
- J. Nielsen and M.H. Caruthers, J. Am. Chem. Soc. 110, 6275–6276 (1988).
- M.A. Dorman, et al., Tetrahedron Symposium in Print, ed., (1984), pp. 95.
- D.J. Dellinger, et al., J. Am. Chem. Soc. 125, 940–950 (2003).
- C.M. Yamada, et al., J. Am. Chem. Soc. 128, 5251–5261 (2006).
- H. Krishna and M.H. Caruthers, J. Am. Chem. Soc. 133, 9844–9854 (2011).
- D.J. Dellinger, et al., J Am. Chem Soc. 133, 11540–11556 (2011).
Page
last modified 19 March 2013.
Copyright © 2003-2013 International Union of Pure and Applied Chemistry.
Questions regarding the website, please contact [email protected]
|





